
EHLERS-DANLOS SİNDROMU
Hüseynova N.M.,
Nağdəliyev Ə.S.,
Abbasova H.N.,
Əliyarzadə T.A.,
Nəbiyeva M.Ə.
ATU-nun Uşaq yoluxucu xəstəlikləri kafedrası,
Respublika Perinatal Mərkəzi
Açar sözlər: sindrom, autosom-dominant, autosom-resessiv, nəsil şəcərəsi, gen mutasiyası, xromosom xəritəsi, Marfanoid habitus, araxnodaktiliya, proband.
Ehlers-Danlos sindromu ilk dəfə 1682-ci ildə Van Meekeren tərəfindən, daha sonralar 1901-ci ildə Ehlers və 1908-ci ildə Danlos tərəfindən təsvir olunmuşdur. Məşhur skripka ustası kimi Paqaninin bu sindromdan əziyyət çəkdiyi ehtimal olunur. Buna sübut kimi müsiqiçinin barmaqlarının qeyri-adi elastikliyi və görünməmiş fitri istedadı göstərilir. Xəstəliyin təsnifatı ilk dəfə Beighton və həmkarları tərəfindən verilmişdir. (6).
Ehlers-Danlos sindromu birləşdirici toxumanın irsi xəstəliyi olub, dərinin, qan damarlarının, oynaqların və daxili orqanların zədələnməsi ilə özünü göstərir.
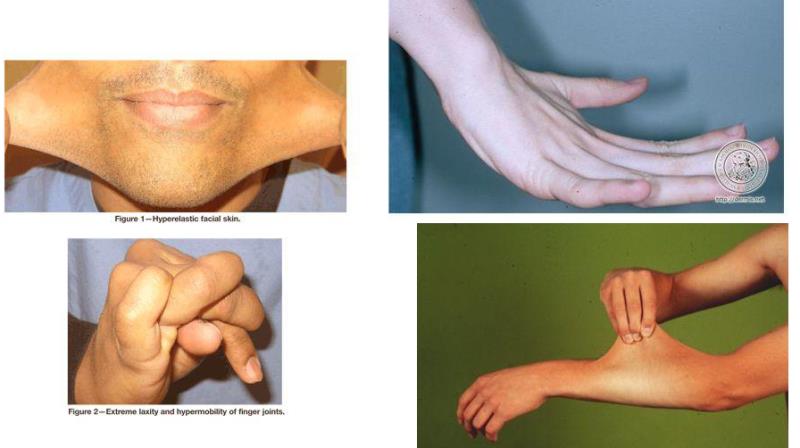
Xəstəlik dərinin hədsiz elastikliyi, oynaqların həddən artıq hərəkətliliyi və toxuma kövrəkliyi ilə xarakterizə olunur. Populyasiyada yayılma tezliyi 1:50000dir (7).
Respublika Perinatal Mərkəzinin (RPM) Tibbi-genetik məsləhət (TGM) kabinetinə fəaliyyətə başladığı gündən etibarən çoxsaylı müraciətlər olmuşdur.
Müraciət edən pasientlər arasında bir çox xromosom və gen patologiyaları aşkar edilmişdir. Bu patologiyalardan biri də Azərbaycanda rəsmi olaraq ilk dəfə qeydə alınan Ehlers-Danlos sindromudur.
Beightonun təsnifatına görə Ehlers-Danlos sindromunun 7 əsas tipii vardır. Hər tipin özünəməxsus byöük və kiçik əlamətləri vardır. Diaqnozu təsdiqləmək üçün heç olmasa bir böyük və bir neçə kiçik əlamətin olması zəruridir.
-
Klassik tip. Bu tipin böyük əlamətlərinə dərinin elastikliyi, geniş atrofik yara çapıqları, oynaqların hipermobilliyi, çıxıqlarlar, pes planus (yastıayaqlılıq) aiddir. Kiçik əlamətlərə isə hamar, sürüşkən məxmərşəkilli dəri, əzələ hipoplaziyası, toxuma kövrəkliyi, yırtıqlar, anal prolaps, servikal çatmamazlıq, mitral qapaq prolapsı, aorta dilatasiyası, hamilələrdə amniotik qişaların vaxtından qabaq cırılması kimi əlamətlər daxildir (9).

Etiologiyası: Xəstəlik autosom-dominant yolla nəsildən nəslə ötürülür. Xəstəliyə səbəb COL5A1 (xromosom xəritəsi 9q34.2-q34.3), COL5A2 (xromosom xəritəsi 2q31) və COL1A1 (xromosom xəritəsi 17q2131-q22) geninin mutasiyasıdır (3).
-
Hipermobil tip (tip III). Bu tipə xas olan böyük əlamətlərə elastik dəri, hipermobil oynaqlar, xüsusilə çiyin, diz, gicgah-çənə oynağının adəti çıxıqları daxildir.
Kiçik əlamətlərə isə xroniki oynaq çıxıqları, ətraflarda ağrılar, müsbət ailə anomnezi, aortanın dilatasiyası, mitral qapaq prolapsı daxil edilir (9).
Etiologiyası: Xəstəlik autosom-dominant yolla nəsildən nəslə ötürülür. Xəstəliyə səbəb COL3A1 və TNXB geninin mutasiyasıdır (3).
-
Vaskulyar tip (tip IV, Arterial – ekximoz)
Etilogiyası: Xəstəliyin ötürülmə tipi autosom-dominant yolladır. Xəstəliyə səbəb COL3A1 geninin mutasiyasıdır. (Xromosom xəritəsi 2q31) (3,4).
Böyük əlamətlərə nazik transluent dəri, bağırsaq və arteriyalarının kövrəkilyi, qanaxmalar, xarakter üz: nazik dimdikşəkilli burun, nazik dodaq, «mələk yanaqları», qabarıq göz aid edilir.
Kiçik əlamətlərə isə xırda oynaqların hipermobilliyi, vətər və əzələ kövrəkliyi, sidik kisəsinin yırtığı, talipes equinovarus (əyripəncəlik), venaların varikozu arterio-venoz fistula, pnevmotoraks, müsbət ailə anamnezi daxildir.
-
Kifoskolioz tipi (tip VI, Ocular skoliotik). Bu tipin böyük əlamətlərinə generalizəolunmuş oynaq labilliyi, yenidoğulmuşun ağır dərəcəli əzələ hipotoniyası və skolioz ilə dünyaya gəlməsi aiddir. Göz skelerasının kövrəkliyi nəticəsində bu xəstələrdə skeleraya qansızmalar da qeydə alınır. Kiçik əlamətlərə toxuma kövrəkliyi, arterial zədələnmə, kiçik zədə nəticəsində əmələ gələn yara və qansızmalar, marfaonid habitus və araxnodaktiliya (hörümçəkşəkilli ətraflar), osteopeniya aiddir (1).

Etiologiyası: Xəstəliyin ötürülmə tipi autosom-resessiv yolla baş verir. Xəstəliyə səbəb kollagen əmələ gətirən lizil-hidroksilaza fermentinin defisitidir ki, bu da PLOD-1 geninin mutasiyası ilə bağlıdır. Bu xəstələr həyatın II-III onilliyində hərəkət qabiliyyətlərini itirə bilərlər (8).
-
Dermatosparaksis tipii (VII C). Böyük əlamətlərə hədsiz dartılan dəri, dərinin hədsiz kövrəkliyi aiddir. Kiçik əlamətlərə misal olaraq quru və boş dərini, kiçik bir zədələnmədən əmələ gələn yaraları, göbək və qasıq yırtığını göstərmək olar.
Etiologiyası: Xəstəlik autosom-resessiv yolla nəsildən nəsilə ötürülür. Xəstəliyə səbəb kimi prokollagen 1N – terminal peptidaza fermentini kodlaşdıran ADAMTS2 geninin mutasiyası göstərilir. (Xromosom xəritəsi 5q23) (8).
-
Artroxalazis tipi (VII A, VII B). böyük əlamətlərə ağır, generalizəolumuş oynaq hipermobilliyi, təkrari çıxıqlar, bud-çanaq oynağının adəti çıxığı aiddir. Kiçik əlamətlərə isə dəri elastikliyi, toxuma kövrəkliyi, kiçik bir zədələnmədən asan əmələ gələn yaralar, əzələ hipotoniyası, kifoskolioz, osteopeniyanı misal göstərmək olar (5).
Etiolologiyası: Xəstərliyin ötürülmə tipi autosom-dominant yolladır. Xəstəliyə səbəb kimi COL1A1, Col1A2 geninin mutasiyası göstərilir. Bu gen kollagen 1-in sintezinə cavabdehdir. (Xromosom xəritəsi 7q22.1). Lakin ədəbiyyata bu əsas tiplərdən başqa digər az yayılmış tiplər də məlumdur (3,8).
Tip V – Ötürülmə tipi X – resessiv yolladır, yəni cinsiyyətlə bağlıdır.
Tip VIII – Ötürülmə tipi autosom-dominant yolladır. Bu tip klassik tipə oxşayır, lakin burada periodontal kövrəklik ön plana çıxır.
Tip X – Ötürülmə tipi autosom-resessiv yolladır. Fibronektin defisiti ilə bağlı trombosit aqreqasiyası ilə xarakterizə olunur (4).
Tip XI – Ötürülmə tipi autosom-dominant yolladır.
Şəxsi müşahidəmiz. RPM-in TGM kabinetinə müraciət edən 8 yaşlı qız uşağında (bundan sonrakı hissələrdə proband adlandırılıcaq) Ehlers-Danlos sindromunun Kifoskolioz tip aşkar edilmişdir. Aşağıda probandan nəsil şəcərəsi nəzərinizə çatdırılır.
Proband birinci hamiləlik, birinci doğuşdandır. Hamiləliyin 41-42-ci həftəsində təbii doğuşla dünyaya gəlib. Anasının deməsinə görə hamiləlik normal keçib. Lakin dölün tərpənməsini çox az və zəif hiss edib. Doğularkən çəkisi 2800 qr, boyu 50 sm olub. Proband ağır dərəəcəli hipotoniya və gözlə görünən skoliozla doğulub. 1 yaşdan sonra oturma vərdişi yaranıb, 1,5 yaşında yanlara çevrilməyə başlayıb, 2,5 yaşında tutaraq gəzməyə başlayıb. Yıxıldıqda yalnız kömək vasitəsi ilə ayağa qalxıb. 3 yaşından sonra sərbəst gəzməyə başlayıb. Anasının sözlərindən belə məlum olur ki, kiçik bir travmadan əmələ gələn yaralar çox gec sağalır. Travma zamanı parçalanan dəri uzun müddət bitişmir, yara açıq qalır. Gec də olsa sağalan yerlərdə isə kobud çapıq toxuması qalır. Göz skelerasında tez-tez qansızmalar olur. Proband uzuq müddət sinir-əzələ xəstəliyi diaqnozu ilə müalicələr alıb: fizioterapiya, massaj, medikamentoz müalicə, ortopedik yardım. Anasının sözlərinə əsasən müalicə fonunda vəziyyəti bir qədər yaxşılaşıb. Bəzi klinik-laborator müayinələrdən keçib. EMQ, qanın biokimyəvi analizi, baş-beyin kompüter-tomoqrafiyası.
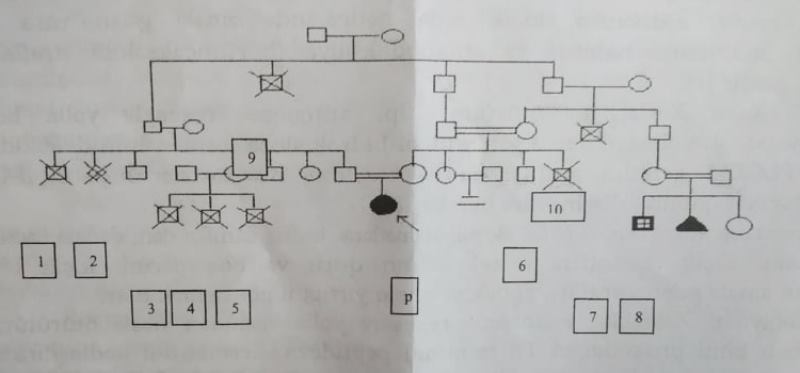
p-proband; 1,2-erkən postnatal ölüm;
3,4,5-erkən neonatal ölüm;
6-gec düşük;
7-lal-kar;
8-erkən düşük;
9-trombemboliya (35 yaş);
10-miokard infarktı (40 yaş).
Obyektiv müayinə: Marfanoid habitus, araznodaktiliya, ağır dərəcəli kifoskolioz və ortopedik korsetin sürtünmə yerləri aydın görünür. Müayinə zamanı oynaqlar hədsiz hipermobildir. Əlin baş barmağı tam arxaya və yana qatlanır. Baş barmaq ovuc üzərinə qoyulanda kənara çıxır. Barmaqlar fleksiya vəziyyətində saidə dəyir. Qulaqlar hərəkətli, göz qapaqları və dirsək dərisi asanlıqla daha uzun məsafəyə dartılır. Proband Ehlers-Danlos sindromuna uyğun demək olar ki, bütün hərəkətlərin öhdəsindən gəlir (dilin ucunu buruna çatdırmaq bacarığından başqa).
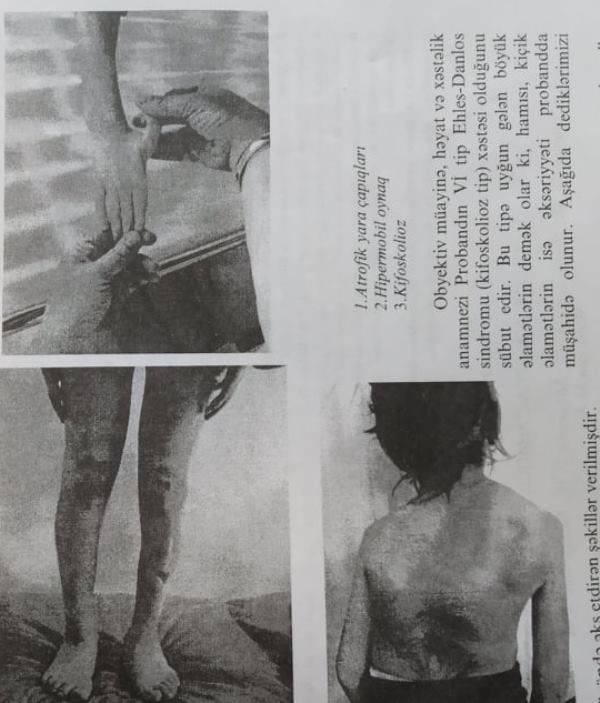
-
Atrofik yara çapıqları
-
Hipermobil oynaq
-
Kifoskolioz
Obyektiv müayinə, həyat və xəstəlik anamnezi Probandan VI tip Ehlers-Danlos sindromu (kifoskolioz tip) xəstəsi olduğunu sübut edir. Bu tipə uyğun gələn böyük əlamətlərin demək olar ki, hamısı, kiçik əlamətlərin isə əksəriyyəti probanda müşahidə olunur. Aşağıda dediklərimizi özündə əks etdirən şəkillər verilmişdir.
Yuxarıda qeyd etdiyimiz kimi sindromun adı çəkilən tipi autosom-resessiv yolla ötürülür. Probandın valideynlərinin ikinci dərəcəli qan qohumu olmaları bu ötürülmə tipini dəstəkləyir. Xəstəlik kollagen sintezini PLOD-1 geni kodlaşdırır. Yəni Ehlers-Danlos sindromunun bu tipi PLOD-1 geninin mutasiyası nəticəsində meydana çıxır.
Diaqnoz sadalanan müayinə metodları sayəsində qoyulur:
-
Dərinin histoloji müayinəsi.
-
Sidikdə lizil-hidroksilaza fermentinin aktivlik dərəcəsinin təyini.
-
Sidikdə deoksipiridinolin: piridinolin nisbətinin təyini. Bu halda nisbət artmış olmalıdır.
-
PLOD-1 geninin molekulyar analizi.
-
Ürəyin exokardioqrafiyası.
Proqnoz: Xəstəlik autosom-resessiv yolla ötürüldüyü üçün növbəti hamiləlikdə təkrarlanma riski 25%-dir (1,2).
Prenatal diaqnostika:
Hamiləliyin 11-13 həftəsində CVS (xorion saçaqlarının biopsiyası) və ya 16-20 həftəsində amniosentez (amniotik mayenin alınması) yolu ilə götürülən materialda PLOD-1 geninin molekulayar analizinin tədqiqi ilə mümkündür. Bu zaman döldə homoziqot mutasiya aşkar olunarsa, nəticə valideynlərə bildirilməlidir. Hamiləliyi pozmaq haqqında qərarı valideynlər birlikdə verməlidirlər (1,2).
Müalicə:
Ehlers-Danlos sindromunun müalicəsi simptomatik və patogenetik üsulla aparılır. Kifoskolioz tipinin əsas madikamentoz müalicəsi askorbin turşusunun yüksək dozları ilə həyata keçirilir. Qeyd etmək lazımdır ki, askorbin turşusu lizil-hidroksilaza fermentinin konfermentidir. Probandan xəstəliyinə səbəb də məhz bu fermentin defisitidir. Digər müalicələrdən əzələlərin ümumi iş qabiliyyətini artırmaq məqsədilə fizioterapiya və massaj tətbiq olur. Yaraların infeksiyalaşmasının profilaktikası məqsədilə antibiotik terapiyası aparılır. Kifoskolioz və pes planusa görə ortopedik vasitələrdən istifadə edilir. Aortal, mitral prolaps zamanı və digər ağır hallarda cərrahi müdaxilə tələb olunur (5).
ƏDƏBİYYAT – LİTERATURA – REFERENCES:
-
Barabas AP. Ehlers-Danlos syndrome; Associated with prematurity and premature rupture of foetal membranes; possible increase in incidence. BMJ2:62,1996
-
Beighton P et al: Revised nosology, Villefranche, 1997. Am J Med Genet 77:31, 1998
-
Burrows NP et al: The molecular genetics of the Ehlers-Danlos syndrome. Clin Exp Dermatol 24:99, 1999.
-
Ehlers E. Cutis laxa, Neigung zu Harmorrhagien in der Haut. 19015. Leier CV et al: The spectrum of cardiac defects in Ehlers-Danlos syndrome, types I and III. 1980
-
Smith’s Recognizable Patterns of Human Malformation. 2006
-
Schievink WI et al: Neurovascular manifestations of heritable disorders of connective tissue. Stroke 25:889,1994
-
Tilistra DJ, Byers PH: Molecular basis of hereditary disorders of connective tissue.
-
Yeowell NH, Pinnell SR: The Ehlers-Danlos syndrome. Semin Dermatol. 1993

WIKIMED.AZ-DA – HƏMİDƏ ABBASOVA HƏKİM GENETİKYOUTUBE VIDEO – PRENATAL DİAQNOSTİKA. TİBBİ-GENETİK MƏSLƏHƏT. HƏKİM-GENETİKİN ROLU. HƏMİDƏ ABBASOVA – HƏKİM-GENETİK
AZƏRBAYCANDA İRSİ-GENETİK XƏSTƏLİKLƏRİN DURUMU HAQQINDA. GENETİK XƏSTƏLİKLƏRLƏ MÜBARİZƏDƏ HƏKİM GENETİKİN ROLU NƏDƏN İBARƏTDİR?
YOUTUBE VİDEO – HƏKİM-GENETİK HƏMİDƏ ABBASOVA: AZƏRBAYCANIN CƏNUB BÖLGƏLƏRİNDƏ, BUZOVNADA BELƏ XƏSTƏLƏR ÇOXDUR…
Facebook Comments