
Leyla Gahramanova, MD, PhD, Tural Galbinur, MD, PhD, DSc, Ayan Mammadkhanova, MD
Background: To report a case of late-onset lysosomal storage disorder presenting with bilateral macular cherry-red spot. Methods: Case report. Results: A 20-year-old female patient with bilateral progressive visual loss was found to have bilateral macular cherry-red spots and was subsequently diagnosed as a possible late-onset Tay–Sachs disease according to results of the genetic analysis. Conclusion: Most lysosomal storage disorders are known as pediatric diseases. However, we must consider the possibility of late-onset lysosomal disorder in case of progressive visual loss and macular cherry-red spot in adult patients. RETINAL CASES & BRIEF REPORTS 00:1–3, 2019
From the *Ophthalmology Division, Caspian International Hospital, Baku, Azerbaijan; and †Department of Ophthalmology, Azerbaijan Medical University, Baku, Azerbaijan. Lysosomal storage disorders (LSDs) are a family of more than 70 rare monogenic diseases that typically present in infancy or childhood and collectively affect 1 in 5,000 live births.1 Lysosomal storage disorders are characterized by the accumulation (so-called “storage”) of nondegraded substrates in the lysosome, with each disease having its own biochemical fingerprint of stored metabolites.2 Cherry-red spot is an ocular manifestation of many inherited lysosomal storage diseases including Tay– Sachs disease, Sandhoff disease, GM1 gangliosidoses, Niemann–Pick disease, Farber disease, metachromatic leukodystrophy, and sialidosis.3
None of the authors has any financial/conflicting interests to disclose. This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-No Derivatives License 4.0 (CCBY-NC-ND), where it is permissible to download and share the work provided it is properly cited. The work cannot be changed in any way or used commercially without permission from the journal. Reprint requests: Tural Galbinur, MD, PhD, DSc, Department of Ophthalmology, Azerbaijan Medical University, Bakikhanov Street 23, Baku-AZ1022, Azerbaijan; e-mail: [email protected]
We report a case of a 20-year-old woman with macular cherry-red spot and late-onset LSD.
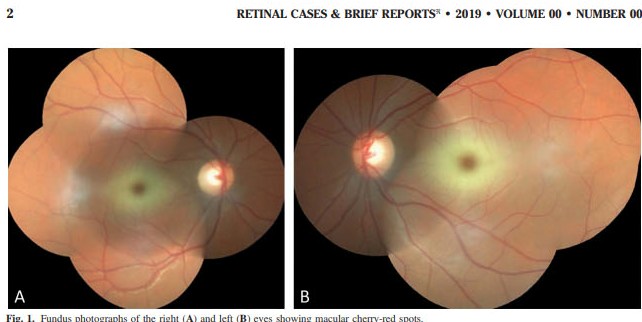
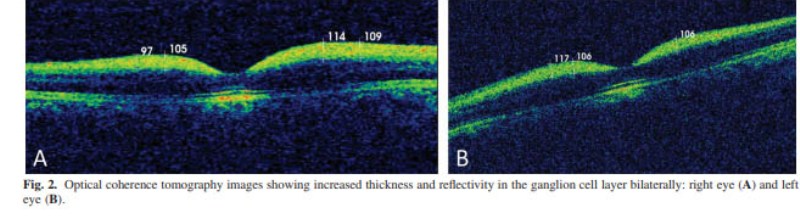
Case Report A 20-year-old female patient presented with progressive visual loss on both eyes to our eye clinic in Baku. She was born of nonconsanguineous parents. Her development was normal in childhood and juvenile period. She had no history of prolonged bleeding due to trauma, hematemesis, fever, night sweats, weight loss, or bone pain. Her parents and two siblings were reportedly normal. On examination, best-corrected visual acuity was 6/9 in the right eye and 6/12 in the left eye. Intraocular pressure was 16 and 14 mmHg in the right eye and the left eye, respectively. Slit-lamp examination of the anterior segment was unremarkable except for blue opacities in both lenses. Dilated fundus examination revealed macular cherry-red spots in both eyes (Figure 1). Optical coherence tomography showed increased thickness and reflectivity in the ganglion cell layer bilaterally indicating the deposition of lipid in the ganglion cells (Figure 2). We referred the patient for abdominal ultrasound examination and magnetic resonance imaging of brain to exclude other organ involvements. Both tests demonstrated normal findings. Patient’s lipid profile was within normal limits. Finally, blood taken for genetic analysis revealed p.Ile436Val (c.1306 A.G) homozygous missense mutation. The missense mutation affected both allele.
Discussion
When evaluating a patient with macular cherry-red spot that is not due to arterial occlusion or trauma, we should consider various forms of LSDs including
Tay–Sachs disease, Sandhoff disease, GM1 gangliosidoses, Niemann–Pick disease, Farber disease, metachromatic leukodystrophy, and sialidosis.3 Most LSDs are known as pediatric diseases, and the adult form of given LSD differs from the childhood disease in several respects. Adult disorders are, with some exceptions, less common than the childhood diseases. The clinical picture is not only less severe, but often shows quite different clinical signs and symptoms than the early-onset form.4 Given the fact that our patient is 20 years old, and she has mild presentation, we considered the different forms of LSDs. We performed abdominal ultrasonography and magnetic resonance imaging of brain to exclude Farber disease, Gaucher disease Type I, Niemann–Pick Type I, gangliosidosis, and metachromatic leukodystrophy. Both tests demonstrated normal findings. Blood tests were within normal limits. Genetic analysis detected a homozygous A.G nucleotide substitution (ATA.GTA) in the nucleotide position 1,306 of the HEXA gene causing the p.Ile436Val (c.1306 A,G) missense mutation. This result indicated a late-onset Tay–Sachs disease or a carrier state to us. Late-onset Tay–Sachs disease is an autosomal recessive LSD due to compound heterozygous or homozygous mutations in HEXA. 5 Patients may present in childhood, adolescence, or early adulthood.6 Although the phenotypic spectrum of the disease has been well characterized, atypical presentations have been described in patients with late-onset Tay–Sachs disease.7 For the time being, treatment options for Tay–Sachs disease and other types of LSDs are very limited. Nevertheless, early diagnosis of the disease is significant as genetic consultation is recommended for the affected families. We also took the patient to the follow-up and offered genetic consultation. This report highlights importance of consideration of LSDs for patients with macular cherry-red spot without typical presentation. Also, it should be noted that early diagnosis of the LSDs is significant to identify the undiagnosed or carrier members of the affected families with genetic consultation. Key words: cherry-red spot, lysosomal storage disease
LATE-ONSET LYSOSOMAL STORAGE DISORDER
References 1. Cox TM, Cachón-González MB. The cellular pathology of lysosomal diseases. J Pathol 2012;226:241–254.
2. Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 2012;199:723–734.
3. Suvarna JC, Hajela SA. Cherry-red spot. J Postgrad Med 2008;54:54–57.
4. Rapola J. Lysosomal storage diseases in adults. Pathol Res Pract 1994;190:759–766.
5. Cao Z, Natowicz MR, Kaback MM, et al. A second mutation associated with apparent beta-hexosaminidase A pseudodeficiency: identification and frequency estimation. Am J Hum Genet 1993;53:1198–1205. 6. Rowland LP. Progressive muscular atrophy and other lower motor neuron syndromes of adults. Muscle Nerve 2010;41:161– 165. 7. Godeiro-Junior C, Felicio AC, Benites V, et al. Late-onset hexosaminidase A deficiency mimicking primary lateral sclerosis. Arq Neuropsiquiatr 2009;67:105–106.

Doctor Tural Galbinur
Doctor Pasha Galninur


Facebook Comments